This forum is intended for questions about kinetics, Surface Plasmon Resonance and the instruments related to these techniques.
No response after the injection of the analyte
- Ran
- Topic Author
- New Member
-

Less
More
- Thank you received: 0
4 years 1 month ago #1
by Ran
No response after the injection of the analyte was created by Ran
Hi everyone,
I met lots of problems with my first SPR experiment and wish I could have someone to help. None of my colleagues have ever worked with SPR before, so I might ask some stupid questions. Sorry about that.
Description: I was using an NTA chip to immobilize a His-tagged protein, and then use DNA aptamer as analytes to test the binding affinity between the DNA aptamer and the protein.
Problem: I saw no response after injecting the DNA aptamer. The response unit is decreasing. However, the reference cell response is flat and equal to 0, so I think it should not be a non-specific binding issue. Also, the DNA aptamers have been reported in the literature with KD around 50 nM. So there should be something wrong with my setup. I hope someone could take a look at my sensorgram and point out which parts are wrong. I feel like some parts are really strange:
1) After the injection of Ni, the bulk effect is large compared with the examples sensorgram on the website. Does that mean I need to further filter my solution? All the buffers are matched.
2) After the injection of protein finished, the signal rapidly decreased, which is much faster than any example sensorgram. Is there anything wrong with this chip? Or I didn't immobilize the protein well on the surface?
3) After the injection of DNA aptamers, I suppose to see a peak but the response unit just kept decreasing. Is there any factor that could cause this result?
Sorry for having soooooo many questions. Any help would be greatly appreciated!
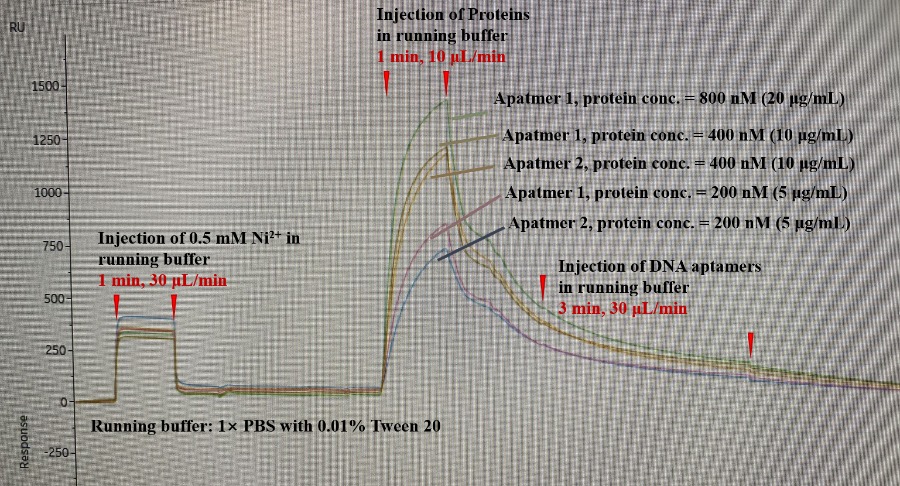
Details:
+ the instrument used: Biacore 8K
+ immobilization conditions:
1) 0.5 mM Ni2+ in running buffer, 30 μL/min for 1 min.
2) wash with 3 mM EDTA in running buffer.
3) 5, 10, 20 μg/mL protein in running buffer, 10 μL/min for 1 min.
+ the buffers and regeneration conditions used:
running buffer: 1×PBS with 0.01% Tween 20.
washing buffer: 1×PBS with 3 mM EDTA, 0.01% Tween 20
regeneration buffer: 1×PBS with 350 mM EDTA, pH=8.4
+ ligand and analyte:
ligand: protein, 24.0 kDa, 5-20 μg/μL (200-800 nM), diluted in the running buffer
analyte: DNA aptamer, 19.0 kDa, 500 nM, diluted in the running buffer
+ Experiment setup:
Conditioning for 1 cycle, Startup for 3 cycles, Analysis for 1 cycle.
I met lots of problems with my first SPR experiment and wish I could have someone to help. None of my colleagues have ever worked with SPR before, so I might ask some stupid questions. Sorry about that.
Description: I was using an NTA chip to immobilize a His-tagged protein, and then use DNA aptamer as analytes to test the binding affinity between the DNA aptamer and the protein.
Problem: I saw no response after injecting the DNA aptamer. The response unit is decreasing. However, the reference cell response is flat and equal to 0, so I think it should not be a non-specific binding issue. Also, the DNA aptamers have been reported in the literature with KD around 50 nM. So there should be something wrong with my setup. I hope someone could take a look at my sensorgram and point out which parts are wrong. I feel like some parts are really strange:
1) After the injection of Ni, the bulk effect is large compared with the examples sensorgram on the website. Does that mean I need to further filter my solution? All the buffers are matched.
2) After the injection of protein finished, the signal rapidly decreased, which is much faster than any example sensorgram. Is there anything wrong with this chip? Or I didn't immobilize the protein well on the surface?
3) After the injection of DNA aptamers, I suppose to see a peak but the response unit just kept decreasing. Is there any factor that could cause this result?
Sorry for having soooooo many questions. Any help would be greatly appreciated!
Details:
+ the instrument used: Biacore 8K
+ immobilization conditions:
1) 0.5 mM Ni2+ in running buffer, 30 μL/min for 1 min.
2) wash with 3 mM EDTA in running buffer.
3) 5, 10, 20 μg/mL protein in running buffer, 10 μL/min for 1 min.
+ the buffers and regeneration conditions used:
running buffer: 1×PBS with 0.01% Tween 20.
washing buffer: 1×PBS with 3 mM EDTA, 0.01% Tween 20
regeneration buffer: 1×PBS with 350 mM EDTA, pH=8.4
+ ligand and analyte:
ligand: protein, 24.0 kDa, 5-20 μg/μL (200-800 nM), diluted in the running buffer
analyte: DNA aptamer, 19.0 kDa, 500 nM, diluted in the running buffer
+ Experiment setup:
Conditioning for 1 cycle, Startup for 3 cycles, Analysis for 1 cycle.
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-

Less
More
- Thank you received: 0
4 years 1 month ago #2
by Arnoud
Replied by Arnoud on topic No response after the injection of the analyte
Hi and welcome.
1) the response level of the Ni2+ could indicate that the concentration is not correct calculated but I am not sure how representative the figure on the website is. Anyhow, this is not really important since you have binding of the HIS-tagged protein.
2) in general, I observe that a higher response on a NTA sensor surface is followed by a higher (initial) dissociation rate. At lower surface saturation the binding looks more stable (due to rebinding?). There seems to be no problem with capturing the HIS-protein.
For difficult interactions, or interactions with low affinity I recommend a different sensor surface (e.g., carboxylated dextran – amine coupling or biotinylated ligand on a streptavidin modified surface). Technical speaking the Ni-HIS interaction is not an immobilization but is considered a capturing of ligand.
3) the fact that the HIS-protein dissociation seems to be uninterrupted upon aptamer injection means that there is no interaction. You state that there should be an interaction. What experiments are done to establish that?
Please refer to this page for more information on aptamers and SPR: www.sprpages.nl/sensor-chips-menu/aptamers
And if you want more information on NTA sensor surface look at www.sprpages.nl/sensor-chips-menu/nta
Kind regards
Arnoud Marquart
1) the response level of the Ni2+ could indicate that the concentration is not correct calculated but I am not sure how representative the figure on the website is. Anyhow, this is not really important since you have binding of the HIS-tagged protein.
2) in general, I observe that a higher response on a NTA sensor surface is followed by a higher (initial) dissociation rate. At lower surface saturation the binding looks more stable (due to rebinding?). There seems to be no problem with capturing the HIS-protein.
For difficult interactions, or interactions with low affinity I recommend a different sensor surface (e.g., carboxylated dextran – amine coupling or biotinylated ligand on a streptavidin modified surface). Technical speaking the Ni-HIS interaction is not an immobilization but is considered a capturing of ligand.
3) the fact that the HIS-protein dissociation seems to be uninterrupted upon aptamer injection means that there is no interaction. You state that there should be an interaction. What experiments are done to establish that?
Please refer to this page for more information on aptamers and SPR: www.sprpages.nl/sensor-chips-menu/aptamers
And if you want more information on NTA sensor surface look at www.sprpages.nl/sensor-chips-menu/nta
Kind regards
Arnoud Marquart
Please Log in or Create an account to join the conversation.
- Ran
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
4 years 1 month ago #3
by Ran
Replied by Ran on topic No response after the injection of the analyte
Hi,
Thank you for your answers! After I read them through, I think the biggest issue I am facing now is that there are not even any changes after I injected the aptamer.
Here I came up with another question: In the ideal case, the capture level should finally keep at a relatively stable level, and the injection of the aptamer should give positive results. However, in my case, the capture signal kept decreasing, so the signal will finally be negative. Is that correct? (I draw a picture in case my description is unclear: ibb.co/93Y5Dwf ) And does that mean I have to change the NTA chip to a real immobilization chip (for example, the carboxylated dextran – amine coupling chip you mentioned)
Since I never really get reliable results, I am just trying to imagine what kind of curve I should finally see. Sorry if I ask any strange questions!
And thank you again for your kind help!
Best wishes,
Thank you for your answers! After I read them through, I think the biggest issue I am facing now is that there are not even any changes after I injected the aptamer.
Here I came up with another question: In the ideal case, the capture level should finally keep at a relatively stable level, and the injection of the aptamer should give positive results. However, in my case, the capture signal kept decreasing, so the signal will finally be negative. Is that correct? (I draw a picture in case my description is unclear: ibb.co/93Y5Dwf ) And does that mean I have to change the NTA chip to a real immobilization chip (for example, the carboxylated dextran – amine coupling chip you mentioned)
Since I never really get reliable results, I am just trying to imagine what kind of curve I should finally see. Sorry if I ask any strange questions!
And thank you again for your kind help!
Best wishes,
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-
Less
More
- Thank you received: 0
4 years 1 month ago - 4 years 1 month ago #4
by Arnoud
Replied by Arnoud on topic No response after the injection of the analyte
The dissociation of the captured ligand can hide some binding. To compensate for this you can subtract a 'blank' - in your case thus a run with the same amount of ligand but without injecting aptamer.
In the second figure the response of the aptamer injection begins after the third red upward arrow and at the first. Even with a decaying surface if there is binding you will see it.
Some literature about decaying surface can be found in
Joss, L., T. A. Morton, M. L. Doyle, et al. - Interpreting kinetic rate constants from optical biosensor data recorded on a decaying surface. Analytical Biochemistry 261: 203-210; (1998).
Di Primo, C. - Real time analysis of the RNAI and RNAII - Rop complex by surface plasmon resonance: from a decaying surface to a standard kinetic analysis. Journal of Molecular Recognition 21: 37-45; (2008).
If a decaying surface is an issue I would recommend a more stable means of immobilization such as a biotin-streptavidin, amine coupling or a high affinity antibody against the ligand.
Technically I don't see any problems so you have to look at your ligand and aptamer if they are really are what you think they are.
Regards
Arnoud
In the second figure the response of the aptamer injection begins after the third red upward arrow and at the first. Even with a decaying surface if there is binding you will see it.
Some literature about decaying surface can be found in
Joss, L., T. A. Morton, M. L. Doyle, et al. - Interpreting kinetic rate constants from optical biosensor data recorded on a decaying surface. Analytical Biochemistry 261: 203-210; (1998).
Di Primo, C. - Real time analysis of the RNAI and RNAII - Rop complex by surface plasmon resonance: from a decaying surface to a standard kinetic analysis. Journal of Molecular Recognition 21: 37-45; (2008).
If a decaying surface is an issue I would recommend a more stable means of immobilization such as a biotin-streptavidin, amine coupling or a high affinity antibody against the ligand.
Technically I don't see any problems so you have to look at your ligand and aptamer if they are really are what you think they are.
Regards
Arnoud
Last edit: 4 years 1 month ago by Arnoud.
Please Log in or Create an account to join the conversation.
Moderators: Arnoud, Arnoud