This forum is intended for questions about kinetics, Surface Plasmon Resonance and the instruments related to these techniques.
Proper Fitting Model
- klevmar
- Topic Author
- New Member
-

Less
More
- Thank you received: 0
9 years 1 week ago #1
by klevmar
Proper Fitting Model was created by klevmar
Hello, I have a question about using the proper fitting model for my data. I'm testing the interaction between a detergent-solubilized receptor and an interacting protein that is fairly large (~80kDa). The receptor forms a homodimer and each subunit should bind one analyte protein, so a 2 analyte per 1 dimer stoichiometry. Previous groups who have analyzed this interaction have used either a bivalent or heterogenous model (most used bivalent) for fitting. In my hands, I can get ok, but not great fits with the bivalent model for one of my receptors, but a close homolog's data will not fit well with that model, but does better with a heterogenous model. It would be fun to use that to speculate on different binding modes between homologs (eg binding of one analyte molecule results in unique conformational changes that affect the binding of the second analyte molecule), but of course I don't want to incorrectly interpret the data. I also created a chimera of the two homologs and analyzed the binding, which also showed a better fit with the heterogenous model, but noticed that KD2 is lower than KD1. Does that mean that the model indicates a stronger affinity for the second analyte occurs after binding of the first?
Here are some additional details that might help determine if I am processing my data correctly:
-I have covalently immobilized an anti-GFP nanobody to a CM5 chip
-I am capturing about 200-250 RU's of a respective GFP-
tagged receptor to the surface
-A concern is that my baseline decreases about 5-10% over the course of an experiment, which I interpret to be a ~10% loss of captured receptor
-Because of my declining baseline, I used the local Rmax parameter in the fittings, which I've provided below
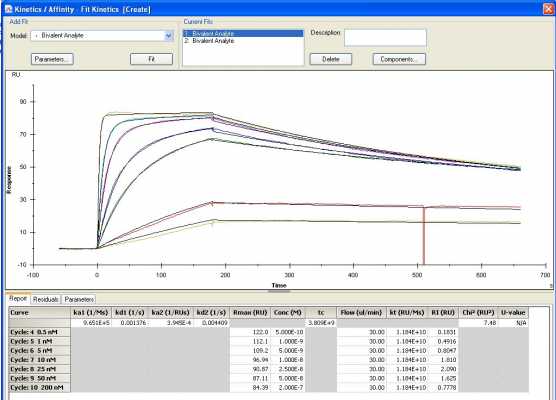
Receptor 1 fit to a bivalent model
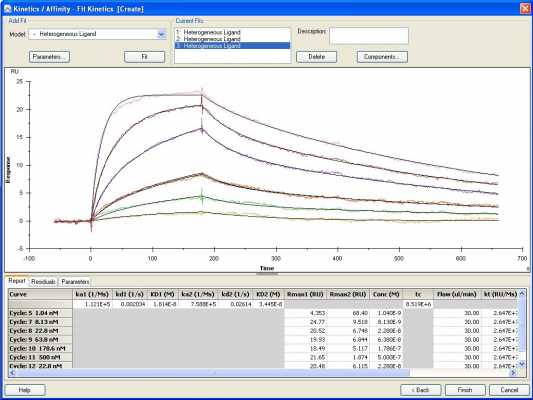
Receptor 2 fit to a heterogenous model
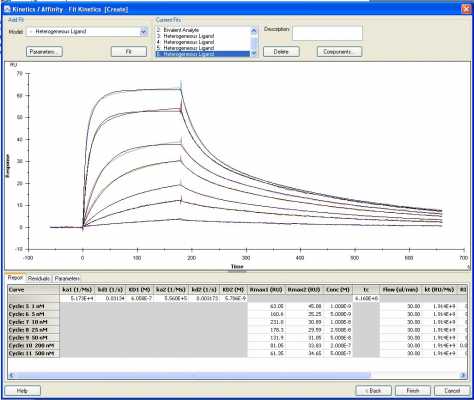
Chimera fit to a heterogenous model
I would greatly appreciate any help you can give!
Here are some additional details that might help determine if I am processing my data correctly:
-I have covalently immobilized an anti-GFP nanobody to a CM5 chip
-I am capturing about 200-250 RU's of a respective GFP-
tagged receptor to the surface
-A concern is that my baseline decreases about 5-10% over the course of an experiment, which I interpret to be a ~10% loss of captured receptor
-Because of my declining baseline, I used the local Rmax parameter in the fittings, which I've provided below
Receptor 1 fit to a bivalent model
Receptor 2 fit to a heterogenous model
Chimera fit to a heterogenous model
I would greatly appreciate any help you can give!
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-

Less
More
- Thank you received: 0
9 years 1 week ago - 9 years 1 week ago #2
by Arnoud
Replied by Arnoud on topic Proper Fitting Model
Hi Klevmar,
First some thoughts about your system and the used models. You say that the receptor dimerizes but both parts bind one analyte molecules. Does one subunit also bind one analyte molecule without being dimerized?
Let’s take a look at the used modes.
A: The bivalent model assumes that the analyte is bivalent, not the ligand. The best known bivalent analyte is of course an antibody. However, when a bivalent molecule is used as a ligand, then the bivalent molecule is treated as having two independent binding sites (figure B ).
The heterogeneous ligand model assumes two independent binding sites on the ligand molecule. Which of the interactions occurs first is dependent on the association rate constants. Independent means that binding to site A is not influencing binding to site B or vice versa.
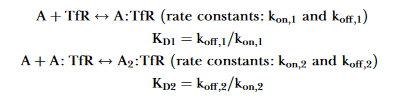
In your case, when two receptor parts form one receptor with two binding sites (C), I would say that there should be a 1:1 binding when both binding sites of the dimerized receptor are independent.
How is a 1:1 fitting of receptor 1 compared to the bivalent analyte fitting?
For the other two fittings it looks as if the Rmax values are a little inconsistent (the numbers are hard to read) over the analyte concentrations. What happens to the fitting when the first Rmax is held constant?
I realize this will not help you much but it is difficult to judge your experiments without more information. Can you share which receptor/analyte you working on or share the publication you refer to?
Kind regards
Arnoud
First some thoughts about your system and the used models. You say that the receptor dimerizes but both parts bind one analyte molecules. Does one subunit also bind one analyte molecule without being dimerized?
Let’s take a look at the used modes.
A: The bivalent model assumes that the analyte is bivalent, not the ligand. The best known bivalent analyte is of course an antibody. However, when a bivalent molecule is used as a ligand, then the bivalent molecule is treated as having two independent binding sites (figure B ).
The heterogeneous ligand model assumes two independent binding sites on the ligand molecule. Which of the interactions occurs first is dependent on the association rate constants. Independent means that binding to site A is not influencing binding to site B or vice versa.
In your case, when two receptor parts form one receptor with two binding sites (C), I would say that there should be a 1:1 binding when both binding sites of the dimerized receptor are independent.
How is a 1:1 fitting of receptor 1 compared to the bivalent analyte fitting?
For the other two fittings it looks as if the Rmax values are a little inconsistent (the numbers are hard to read) over the analyte concentrations. What happens to the fitting when the first Rmax is held constant?
I realize this will not help you much but it is difficult to judge your experiments without more information. Can you share which receptor/analyte you working on or share the publication you refer to?
Kind regards
Arnoud
Last edit: 9 years 1 week ago by Arnoud.
Please Log in or Create an account to join the conversation.
- klevmar
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
9 years 6 days ago - 9 years 6 days ago #3
by klevmar
Replied by klevmar on topic Proper Fitting Model
Thank you for taking the time to reply, I really appreciate it as I'm the only one doing SPR work here and am still learning. The receptor is transferrin receptor 1 (though I'm also looking at TFR2) and the analyte is transferrin. It's been characterized by SPR several times and here is one example article where they used the bivalent model (
journals.plos.org/plosbiology/article?id...rnal.pbio.0000051#s4
). I believe their assertion is that the binding is sequential, or that the binding to one subunit affects the binding of the other. My system is with the full-length protein rather than truncated binding domain, but should roughly follow their "Wild-type" and holo-TF results at pH 7.4.
To answer the dimerization question, they should all be in the dimer form and not dissociating to monomer. I am isolating the dimer species by size exclusion and the dimer forms multiple cross-subunit disulfides to stay in that form.
Thanks for the information on the different models. The 1:1 model It doesn't fit very well at either association or dissociation phases for the the receptors. I've played around with Rmax and RI parameters a bit with no significant improvement.
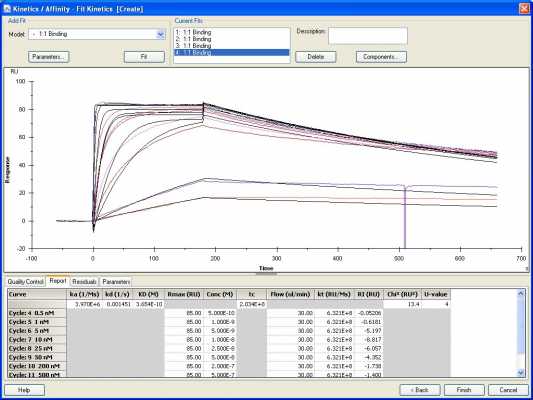
Here is the TfR1 1:1 fitting with a constant Rmax (set at 85).
The Rmax values are inconsistent when I do set a local Rmax parameter (for TfR1 bivalent, they range from 85-122 RU's). Would this possibly be from loss of receptor over the course of an experiment?
Please let me know if you could use any further information to help. I really appreciate it.
To answer the dimerization question, they should all be in the dimer form and not dissociating to monomer. I am isolating the dimer species by size exclusion and the dimer forms multiple cross-subunit disulfides to stay in that form.
Thanks for the information on the different models. The 1:1 model It doesn't fit very well at either association or dissociation phases for the the receptors. I've played around with Rmax and RI parameters a bit with no significant improvement.
Here is the TfR1 1:1 fitting with a constant Rmax (set at 85).
The Rmax values are inconsistent when I do set a local Rmax parameter (for TfR1 bivalent, they range from 85-122 RU's). Would this possibly be from loss of receptor over the course of an experiment?
Please let me know if you could use any further information to help. I really appreciate it.
Last edit: 9 years 6 days ago by klevmar.
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-
Less
More
- Thank you received: 0
9 years 5 days ago #4
by Arnoud
Replied by Arnoud on topic Proper Fitting Model
HI,
First an answer to your questions
After reading the article I understand better now. Correct me when I am wrong but you capture the TfR receptor (a homodimer) and the analyte are homologs to Fe-Tf (monomers).
In the publication is shown that the interaction is:
Each binding of A gives a response. In the case of a bivalent model, only the first interaction generates response and the second interaction will lower the overall binding strength (Did you notice that the second ka is in RU-1 s-1?).
From the interaction above I would choose for the heterogeneous model (independent sites). If the first interaction changes the second binding site is hard to predict and you cannot prove that by using a different analysis model.
One other thing you can do is to inject the analyte a little longer until all concentrations reach equilibrium and do an equilibrium analysis. Although you won’t have the rate constants you probably can extract one perhaps two equilibrium dissociation constants.
www.sprpages.nl/data-fitting/models/equilibrium-analysis
First an answer to your questions
If the Rmax is decreasing with each cycle it probably is.The Rmax values are inconsistent when I do set a local Rmax parameter (for TfR1 bivalent, they range from 85-122 RU's). Would this possibly be from loss of receptor over the course of an experiment?
With models that have more than one interaction set you have to be careful because between fittings, the values of the kinetic parameters can switch. Thus ka1 and kd1 can become ka2 and kd2 in the next fitting (likewise for Rmax). Second problem is that the model does not know which interaction comes first. It is assumed that the interaction with the highest ka occurs first, but I have never seen prove of that. I don’t know if someone did some simulations or modelling on this.I also created a chimera of the two homologs and analyzed the binding, which also showed a better fit with the heterogeneous model, but noticed that KD2 is lower than KD1. Does that mean that the model indicates a stronger affinity for the second analyte occurs after binding of the first?
After reading the article I understand better now. Correct me when I am wrong but you capture the TfR receptor (a homodimer) and the analyte are homologs to Fe-Tf (monomers).
In the publication is shown that the interaction is:
Attachment klevmar.png not found
Each binding of A gives a response. In the case of a bivalent model, only the first interaction generates response and the second interaction will lower the overall binding strength (Did you notice that the second ka is in RU-1 s-1?).
From the interaction above I would choose for the heterogeneous model (independent sites). If the first interaction changes the second binding site is hard to predict and you cannot prove that by using a different analysis model.
One other thing you can do is to inject the analyte a little longer until all concentrations reach equilibrium and do an equilibrium analysis. Although you won’t have the rate constants you probably can extract one perhaps two equilibrium dissociation constants.
www.sprpages.nl/data-fitting/models/equilibrium-analysis
Attachments:
Please Log in or Create an account to join the conversation.
Moderators: Arnoud, Arnoud