This forum is intended for questions about kinetics, Surface Plasmon Resonance and the instruments related to these techniques.
big drop at the start of dissociation
- lee
- Topic Author
- New Member
-

Less
More
- Thank you received: 0
8 years 1 month ago #1
by lee
big drop at the start of dissociation was created by lee
Dear SPR community,
I am using SPR to study interactions between proteins and peptides. Recently, I have some data that i could not make sense of so I am hoping someone might have any idea or experiences on similar things.
I ran the SPR experiments with BIAcore T200. My his-tagged protein was capture onto a NTA chip and the analyte (peptide) was diluted in the same buffer as my SPR running buffer. The running buffer is similar to a standard 1x HBS-N buffer with 0.1% detergent.
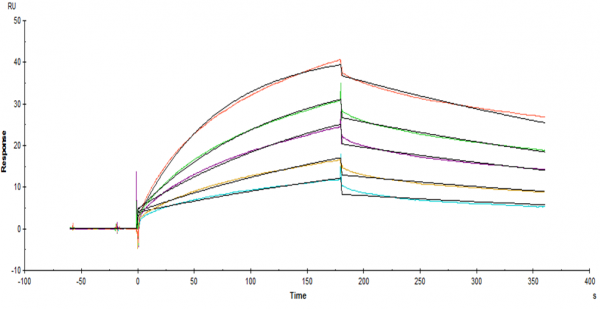
The sensorgrams given here are the three experiments i did for three different peptides under the same conditions. What i do not understand is that why both figure 1 and 2 have a big drop at the start of the dissociation. Could it be the differences in buffer? (But figure 3 is fine) Does anyone know what might have happened to cause this phenomena.
thank you so much for your help
figure 1
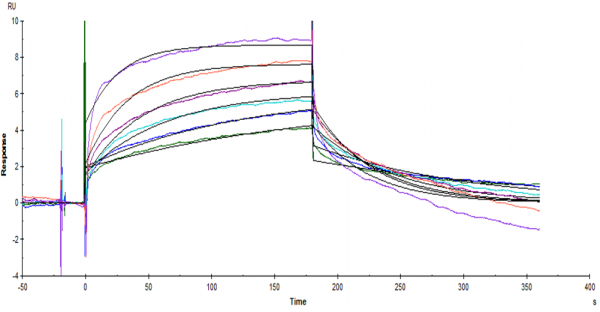
figure 2
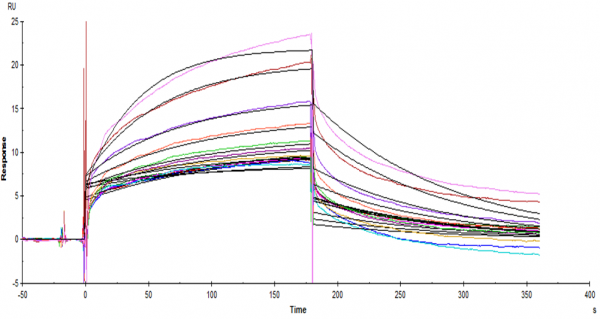
figure 3
I am using SPR to study interactions between proteins and peptides. Recently, I have some data that i could not make sense of so I am hoping someone might have any idea or experiences on similar things.
I ran the SPR experiments with BIAcore T200. My his-tagged protein was capture onto a NTA chip and the analyte (peptide) was diluted in the same buffer as my SPR running buffer. The running buffer is similar to a standard 1x HBS-N buffer with 0.1% detergent.
The sensorgrams given here are the three experiments i did for three different peptides under the same conditions. What i do not understand is that why both figure 1 and 2 have a big drop at the start of the dissociation. Could it be the differences in buffer? (But figure 3 is fine) Does anyone know what might have happened to cause this phenomena.
thank you so much for your help
figure 1
figure 2
figure 3
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-

Less
More
- Thank you received: 0
8 years 1 month ago #2
by Arnoud
Replied by Arnoud on topic big drop at the start of dissociation
Hi Lee,
It is not only the drop at the start of the dissociation. The same is happening at the start of the association (jump-up). Even when the buffer is matched your peptide can have some bulk-effect. The bulk effect by the peptide is concentration dependent (high with high concentrations).P
Peptides are mostly used in high concentration (µM range) so you can expect this.
Did you compare the raw reference and active channel? Probably there is a difference in the bulk jump which is causing the jump in the subtracted curves.
Not so easy to solve though. Somehow you have to change the reference channel to mimic the same bulk behaviour to the injected peptide concentration. If you use a deactivated channel, immobilizing a protein can do the trick.
As you noticed in figure 1 and 2, the fitting is not following the curves. The algorithm tries to make the fit better by adding a buffer jump. If the jump is small (and proportional to the analyte concentration) this should not be a problem but with figure 1 and 2 the fit result is not reliable.
Hope you can solve the difference. Let us know.
Kind regards
Arnoud
It is not only the drop at the start of the dissociation. The same is happening at the start of the association (jump-up). Even when the buffer is matched your peptide can have some bulk-effect. The bulk effect by the peptide is concentration dependent (high with high concentrations).P
Peptides are mostly used in high concentration (µM range) so you can expect this.
Did you compare the raw reference and active channel? Probably there is a difference in the bulk jump which is causing the jump in the subtracted curves.
Not so easy to solve though. Somehow you have to change the reference channel to mimic the same bulk behaviour to the injected peptide concentration. If you use a deactivated channel, immobilizing a protein can do the trick.
As you noticed in figure 1 and 2, the fitting is not following the curves. The algorithm tries to make the fit better by adding a buffer jump. If the jump is small (and proportional to the analyte concentration) this should not be a problem but with figure 1 and 2 the fit result is not reliable.
Hope you can solve the difference. Let us know.
Kind regards
Arnoud
Please Log in or Create an account to join the conversation.
- lee
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
8 years 1 month ago #3
by lee
Replied by lee on topic big drop at the start of dissociation
Hi Arnoud,
thank you very much for your help.
I checked the raw data and you were right. there is a clear difference between the curves and that differences get bigger and bigger as the peptide concentration increases.
Do you think it is possible and reasonable that this bulk effect is peptide dependent? because the peptide i use in figure 3 has similar concentrations but the bulk effect is very small.
regards
lee
thank you very much for your help.
I checked the raw data and you were right. there is a clear difference between the curves and that differences get bigger and bigger as the peptide concentration increases.
Do you think it is possible and reasonable that this bulk effect is peptide dependent? because the peptide i use in figure 3 has similar concentrations but the bulk effect is very small.
regards
lee
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-
Less
More
- Thank you received: 0
8 years 1 month ago #4
by Arnoud
Replied by Arnoud on topic big drop at the start of dissociation
Hi Lee,
Yes I think it is peptide dependent. Why? I think the difference in peptide composition/length and possible contaminant from the synthesis.
In addition, the volume you use to dissolve can matter.
We normally make a stock solution of about 1 mg/ml but the volume of water (or buffer) to add is different depending on the peptide. Hence you get some solution (bulk) differences.
Kind regards
Arnoud
Yes I think it is peptide dependent. Why? I think the difference in peptide composition/length and possible contaminant from the synthesis.
In addition, the volume you use to dissolve can matter.
We normally make a stock solution of about 1 mg/ml but the volume of water (or buffer) to add is different depending on the peptide. Hence you get some solution (bulk) differences.
Kind regards
Arnoud
Please Log in or Create an account to join the conversation.
Moderators: Arnoud, Arnoud