This forum is intended for questions about kinetics, Surface Plasmon Resonance and the instruments related to these techniques.
Which method reduces negative curve by buffer conc. mismatch?
- Koreanraichu
- Topic Author
- New Member
-

Less
More
- Thank you received: 0
8 years 3 months ago #1
by Koreanraichu
Which method reduces negative curve by buffer conc. mismatch? was created by Koreanraichu
Greetings, I am SPR newbie whi live in KOR, and I need your help.
recently, I went my home at 11:00 p.m because same experiment, and it ruind by negative curve by buffer conc. mismatch. I'm soooooo exhausted...
I think, this negative curve occurs by dilution method. So you just say what's the problem and say prefer method.
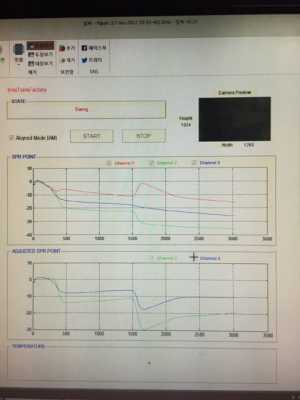
Method 1(run at 17th Jan)
for 1x TBS w/sample
10x TBS 100ul
sample 100ul
DW 800ul
it's my co-worker's opinion. He said "DW stock in diluted at dH2O(DW), and 10x sample was diluted at DW, so you match concentration TBS with 1x TBS buffer. If you dilute sample at 1x TBS, true conc of TBS in sample will be 0.9x."
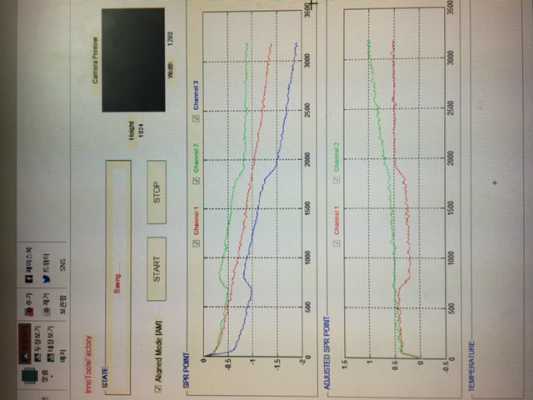
Method 2(27th, Dec 2016)
sample 100ul(serial diluted at 1X TBS, stock is 1000X)
1x TBS 900ul
it's my opinion which counter argument of co-worker's opinion.
"Then, how about serial dilution at 1X TBS? stock solution is diluted in DW because we don't know what buffer uses in analysis this. If we do serial dilution, this stock is 1000X then true conc will be 0.999X, same as 1X TBS for washing. "
he agreed with my opinion, but he changed attitude when result is bad...
sample is 5bp long DNA which have differ 1bp in its chain.
recently, I went my home at 11:00 p.m because same experiment, and it ruind by negative curve by buffer conc. mismatch. I'm soooooo exhausted...
I think, this negative curve occurs by dilution method. So you just say what's the problem and say prefer method.
Method 1(run at 17th Jan)
for 1x TBS w/sample
10x TBS 100ul
sample 100ul
DW 800ul
it's my co-worker's opinion. He said "DW stock in diluted at dH2O(DW), and 10x sample was diluted at DW, so you match concentration TBS with 1x TBS buffer. If you dilute sample at 1x TBS, true conc of TBS in sample will be 0.9x."
Method 2(27th, Dec 2016)
sample 100ul(serial diluted at 1X TBS, stock is 1000X)
1x TBS 900ul
it's my opinion which counter argument of co-worker's opinion.
"Then, how about serial dilution at 1X TBS? stock solution is diluted in DW because we don't know what buffer uses in analysis this. If we do serial dilution, this stock is 1000X then true conc will be 0.999X, same as 1X TBS for washing. "
he agreed with my opinion, but he changed attitude when result is bad...
sample is 5bp long DNA which have differ 1bp in its chain.
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-

Less
More
- Thank you received: 0
8 years 3 months ago #2
by Arnoud
Replied by Arnoud on topic Which method reduces negative curve by buffer conc. mismatch?
Hi, welcome to the SPR pages.
If I look at your sensorgrams is see a lot of drift in the lines. First step is to equilibrate the system in order to have nice stable lines to begin with.
The other strange thing is, the (buffer) jump occur only with two of the three lines. Is the analyte flowing over all three channels and is the red one the reference? A buffer mismatch between sample and running buffer should be visible in all channels where the analyte is flowing over.
Minimizing buffer mismatch is best done by dialysing the analyte against the flow buffer. When this is not possible, try to match the flow buffer by using the flow buffer in all dilution steps. You can try to dilute the analyte in such a way that the end concentration of the buffer components is equal to the flow buffer. However, in the Biacore systems, 1 mM of NaCl difference between analyte and flow buffer can have a response of 20 RU.
What the best strategy to dilute your analyte also depends on the concentration of the analyte and the buffer. We prefer high concentration analytes preferably in running buffer. This gives a good opportunity to dilute the analyte 100 – 100 times. With low analyte concentrations, buffer mismatch will be more extensive, but with proper referencing this can be overcome.
For the dilution scheme I would make dilution steps in running buffer. Trying to match the running buffer out of 10x stocks is more difficult.
Kind regards
Arnoud
If I look at your sensorgrams is see a lot of drift in the lines. First step is to equilibrate the system in order to have nice stable lines to begin with.
The other strange thing is, the (buffer) jump occur only with two of the three lines. Is the analyte flowing over all three channels and is the red one the reference? A buffer mismatch between sample and running buffer should be visible in all channels where the analyte is flowing over.
Minimizing buffer mismatch is best done by dialysing the analyte against the flow buffer. When this is not possible, try to match the flow buffer by using the flow buffer in all dilution steps. You can try to dilute the analyte in such a way that the end concentration of the buffer components is equal to the flow buffer. However, in the Biacore systems, 1 mM of NaCl difference between analyte and flow buffer can have a response of 20 RU.
What the best strategy to dilute your analyte also depends on the concentration of the analyte and the buffer. We prefer high concentration analytes preferably in running buffer. This gives a good opportunity to dilute the analyte 100 – 100 times. With low analyte concentrations, buffer mismatch will be more extensive, but with proper referencing this can be overcome.
For the dilution scheme I would make dilution steps in running buffer. Trying to match the running buffer out of 10x stocks is more difficult.
Kind regards
Arnoud
Please Log in or Create an account to join the conversation.
- Koreanraichu
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
8 years 3 months ago - 8 years 3 months ago #3
by Koreanraichu
Replied by Koreanraichu on topic Which method reduces negative curve by buffer conc. mismatch?
1. one line is reference channel then it flow only running buffer. (red line in 2nd picture) maybe in 1st pic, reference channel is blue line. (two sample+one reference)
2. you mean Serial dilution by 1X TBS > 10X TBS+(DNA+DW)? I disputing about dilution method with co-worker...
3. I use ITF-300(innotools factory). it can detect sample 100pg(or 100pM), then sample conc.is sufficient(100nM).
4. sample is 5bp DNA. I think it is so hard to detect, me and co-worker want to see it can detect 1bp difference in 5bp length DNA.
2. you mean Serial dilution by 1X TBS > 10X TBS+(DNA+DW)? I disputing about dilution method with co-worker...
3. I use ITF-300(innotools factory). it can detect sample 100pg(or 100pM), then sample conc.is sufficient(100nM).
4. sample is 5bp DNA. I think it is so hard to detect, me and co-worker want to see it can detect 1bp difference in 5bp length DNA.
Last edit: 8 years 3 months ago by Koreanraichu.
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-
Less
More
- Thank you received: 0
8 years 3 months ago - 8 years 3 months ago #4
by Arnoud
Replied by Arnoud on topic Which method reduces negative curve by buffer conc. mismatch?
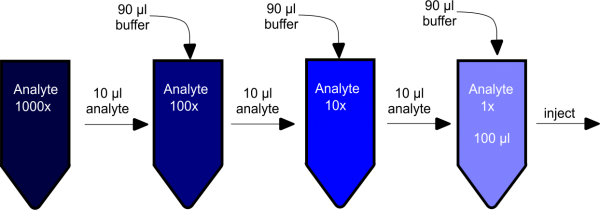
The best dilution option (in my opinion) is to dilute the analyte directly into the running buffer. If I understand correctly, your stock solution is 1000x. Thus I would dilute 10 µl of stock (1000x) + 90 µl of flow buffer (1x TBS in your case). Then 10 µl (100x) + 90µl and 10 µl (10x) + 90 µl giving you 100 µl 1x analyte in almost 100% flow buffer (adjust the volumes at your convenience).
There must be some literature about DNA / oligo’s and mutational detection. Maybe I can find some references for you.
Kind regards
Arnoud
There must be some literature about DNA / oligo’s and mutational detection. Maybe I can find some references for you.
Kind regards
Arnoud
Last edit: 8 years 3 months ago by Arnoud. Reason: typo
Please Log in or Create an account to join the conversation.
- Koreanraichu
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
8 years 3 months ago #5
by Koreanraichu
Replied by Koreanraichu on topic Which method reduces negative curve by buffer conc. mismatch?
I would dilute 10 µl of stock (1000x) + 90 µl of flow buffer (1x TBS in your case). Then 10 µl (100x) + 90µl and 10 µl (10x) + 90 µl giving you 100 µl 1x analyte in almost 100% flow buffer
>>this in total vol. 1ml(1000ul)?
>>this in total vol. 1ml(1000ul)?
Please Log in or Create an account to join the conversation.
- Arnoud
- Moderator
-
Less
More
- Thank you received: 0
8 years 3 months ago #6
by Arnoud
Replied by Arnoud on topic Which method reduces negative curve by buffer conc. mismatch?
I would follow the dilution scheme depicted. The example has a 10x dilution step and the final volume of the 1x solution is 100 µl. But if you take 100 µl analyte + 900 buffer in each step your final solution volume is 1 ml with 1x analyte concentration.
Please Log in or Create an account to join the conversation.
Moderators: Arnoud, Arnoud